La consulta por un paciente pediátrico de 2 años de edad o algo más por transaminasas elevadas o moderadamente elevadas, suele ser frecuente y en ocasiones la evolución posterior de estos pacientes puede plantear problemas.
El pediatra descartará una serie de procesos como hepatitis B o C u otras hepatitis virales, procesos metabólicos afectando al hígado, deficiencia de alfa 1 antitripsina, galactosemia, fructosemia, trastornos de la beta oxidación, trastornos del ciclo de la urea, hepatitis autoinmunes o bien la causa de las transaminasas elevadas puede ser muscular. Raramente a esa edad, por debajo de 5 años o menos, pensamos en la enfermedad de Wilson.
Recordemos brevemente que es la Enfermedad de Wilson (Medline plus):
Es un trastorno hereditario que causa que haya demasiado cobre en los tejidos del cuerpo. El exceso de cobre causa daño al hígado y al sistema nervioso; es un trastorno hereditario poco común. En caso de que ambos padres sean portadores de un gen anormal para la enfermedad de Wilson, hay un 25% de probabilidades en cada embarazo de que el niño sufra el trastorno y que el cuerpo absorba y conserve demasiado cobre. Este cobre se deposita en el hígado, el cerebro, los riñones y los ojos. Los depósitos de cobre ocasionan daño y muerte tisular y cicatrización, lo cual hace que los órganos afectados dejen de funcionar bien.
Esta enfermedad es más común en personas de Europa oriental, Sicilia y el sur de Italia, aunque se puede presentar en cualquier grupo. La enfermedad de Wilson aparece típicamente en personas menores de 40 años. En los niños, los síntomas comienzan a aparecer alrededor de los 5 años o antes
Los síntomas y signos pueden incluir simple elevación de las tansminasas precozmente y en estadíos más avanzados: postura anormal de brazos y piernas, c
onfusión o
delirio, d
emencia, dificultad y rigidez para mover los brazos y las piernas, d
ificultad para caminar (ataxia), cambios emocionales o conductuales, agrandamiento del abdomen (
distensión abdominal), cambios de personalidad, f
obias, angustia (neurosis), m
ovimientos lentos, lentitud o disminución de los movimientos y expresiones faciales, deterioro del lenguaje , t
emblores en los brazos o en las manos, m
ovimientos incontrolables, m
ovimientos impredecibles o espasmódicos, v
ómito con sangre, d
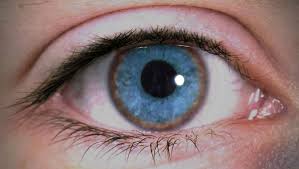
ebilidad, piel amarilla (ictericia) o color amarillo de la esclerótica del ojo (ictericia). Presencia del anillo corneal de Kayser Fleischer
Recordemos que el los alimentos que llevan más cobre son las vísceras, mariscos, chocolate, ciertas aguas, o el uso de vasijas o cañerías que desprendan cobre...Algunos de estos alimentos es raro que sean ingeridos por niños de corta edad, pero a pesar de ello debemos pensar ante unas transaminasas elevadas aún por debajo de los 5 años, en esta enfermedad.
Por ello es interesante que leais el articulo del JPGN sobre la Enfermedadde Wilson en pequeñines o bien este resumen:
Comienzo temprano de la enfermedad de Wilson - desafíos diagnósticos .Wiernicka, Anna; Dadalski, Maciej; Janczyk, Wojciech; Kaminska, Diana;Naorniakowska, Magdalena; Hüsing-Kabar, Anna; Schmidt, Hartmut; Socha, Piotr . (C) 2017 by European Society for Pediatric Gastroenterology, Hepatology, and Nutrition and North American Society for Pediatric Gastroenterology, JPGN, julio de 2017
Objetivos: Analizar las presentaciones clínicas, el diagnóstico y el tratamiento de pacientes con edades por debajo de 5 años con enfermedad de Wilson temprana (WD).
Métodos: Los datos de 143 pacientes pediátricos con WD tratada en nuestro centro entre enero de 1996 y noviembre de 2015 se analizaron retrospectivamente.
Resultados: Una revisión de los 143 pacientes pediátricos con WD puso de manifiesto 21 (10 niñas, 11 niños) con unos primeros síntomas o resultados anormales de las pruebas de función hepática a la edad de 5 años o por debajo de los 5 años. El diagnóstico de WD fue confirmado en ocho pacientes menores de 5 años. Al inicio del estudio, el nivel medio de alanina aminotransferasa en suero fue de 222 U / ly el nivel medio de aspartato aminotransferasa en suero fue de 130 U / l, La concentración media de ceruloplasmina sérica en 16 pacientes evaluados fue por debajo de 20 mg / dl. De los 15 pacientes que se sometieron a pruebas de excreción urinaria de cobre, ocho tenían niveles entre 40 y 100 μ g / día, con sólo cuatro con niveles por encima de 100 μ g / día. La cuantificación de cobre de hígado fue superior a 250 μ g / g de peso seco en 16 pacientes. La mutación más común fue p.H1069Q, con heterozigosis compuesta en cinco pacientes y homocigosis en nueve.Dieciséis pacientes fueron tratados con sales de zinc y cinco con D-penicilamina (Cupripen).Ambos tratamientos fueron eficaces, sin efectos secundarios graves observados después de 3-24 meses.
Conclusiones: La enfermedad de Wilson puede presentarse ya a los 2 años de edad. Debido a que las pruebas bioquímicas pueden ser menos sensibles en niños muy pequeños, los diagnósticos pueden requerir una combinación de pruebas. Si las pruebas moleculares no son concluyentes, se debe practicar una biopsia hepática y medir el contenido de cobre en el hígado.
(C) 2017 por la Sociedad Europea de Gastroenterología Pediátrica, Hepatología y Nutrición y la Sociedad Norteamericana de Gastroenterología Pediátrica
Nota adicional del Dr Tormo:
es conveniente una vez iniciado el tratamiento con D- Penicilamima, no interrumpirlo, para realizar la prueba de verificación de menor eliminación de cobre por orina, tras suprimir la D-Penicilamina, por el peligro de la presentación de un coma hepático agudo.
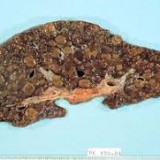
Es una enfermedad a diagnósticar precozmente o puede acabar en cirrosis, hipertensión portal o en el mejor de los casos en trasplante de hígado; en cambio con dieta y tratamiento adecuado, no va a plantear ningún problema.